Activity 4: From single scattering to multiple scattering#
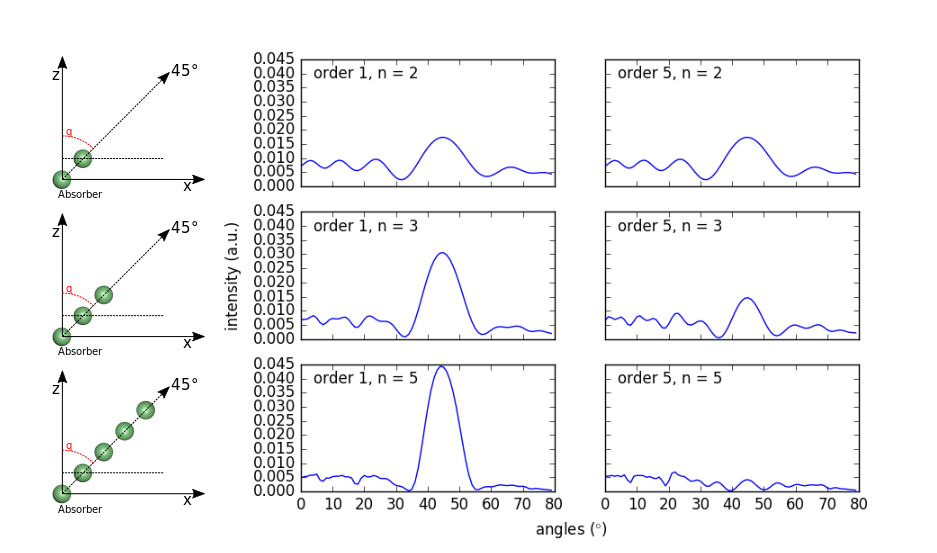
Fig. 10 Polar scan of a Ni chain of 2-5 atoms for single and mutliple (5th order) scattering.#
See also
based on this paper from M.-L. Xu et al. Phys. Rev. B 39 p8275 (1989)
1# coding: utf-8
2
3# import all we need and start by msspec
4from msspec.calculator import MSSPEC
5
6# we will build a simple atomic chain
7from ase import Atom, Atoms
8
9# we need some numpy functions
10import numpy as np
11
12
13symbol = 'Ni' # The kind of atom for the chain
14orders = (1, 5) # We will run the calculation for single scattering
15 # and multiple scattering (5th diffusion order)
16chain_lengths = (2,3,5) # We will run the calculation for differnt lengths
17 # of the atomic chain
18a = 3.499 * np.sqrt(2)/2 # The distance bewteen 2 atoms
19
20# Define an empty variable to store all the results
21all_data = None
22
23# 2 for nested loops over the chain length and the order of diffusion
24for chain_length in chain_lengths:
25 for order in orders:
26 # We build the atomic chain by
27 # 1- stacking each atom one by one along the z axis
28 chain = Atoms([Atom(symbol, position = (0., 0., i*a)) for i in
29 range(chain_length)])
30 # 2- rotating the chain by 45 degrees with respect to the y axis
31 #chain.rotate('y', np.radians(45.))
32 chain.rotate(45., 'y')
33 # 3- setting a custom Muffin-tin radius of 1.5 angstroms for all
34 # atoms (needed if you want to enlarge the distance between
35 # the atoms while keeping the radius constant)
36 #[atom.set('mt_radius', 1.5) for atom in chain]
37 # 4- defining the absorber to be the first atom in the chain at
38 # x = y = z = 0
39 chain.absorber = 0
40
41 # We define a new PED calculator
42 calc = MSSPEC(spectroscopy = 'PED')
43 calc.set_atoms(chain)
44 # Here is how to tweak the scattering order
45 calc.calculation_parameters.scattering_order = order
46 # This line below is where we actually run the calculation
47 all_data = calc.get_theta_scan(level='3s', #kinetic_energy=1000.,
48 theta=np.arange(0., 80.), data=all_data)
49
50 # OPTIONAL, to improve the display of the data we will change the dataset
51 # default title as well as the plot title
52 t = "order {:d}, n = {:d}".format(order, chain_length) # A useful title
53 dset = all_data[-1] # get the last dataset
54 dset.title = t # change its title
55 # get its last view (there is only one defined for each dataset)
56 v = dset.views()[-1]
57 v.set_plot_options(title=t) # change the title of the figure
58
59
60
61# OPTIONAL, set the same scale for all plots
62# 1. iterate over all computed cross_sections to find the absolute minimum and
63# maximum of the data
64min_cs = max_cs = 0
65for dset in all_data:
66 min_cs = min(min_cs, np.min(dset.cross_section))
67 max_cs = max(max_cs, np.max(dset.cross_section))
68
69# 2. for each view in each dataset, change the scale accordingly
70for dset in all_data:
71 v = dset.views()[-1]
72 v.set_plot_options(ylim=[min_cs, max_cs])
73
74# Pop up the graphical window
75all_data.view()
76# You can end your script with the line below to remove the temporary
77# folder needed for the calculation
78calc.shutdown()
Some questions to answer