Activity 3: Adsorbates and the single scattering approach#
Photoelectron diffraction is widely used to study the adsorption of atoms or molecules on a crystalline surface. Photoelectrons from adsorbates are scattered by the underlying surface, carrying information about the adsorption site, bond length and/or molecule orientation…. Thanks to a simulation, such information becomes quantitative with a high degree of accuracy.
Calculations of the multiple scattering using matrix inversion have the great advantage of being exact, including all scattering paths. On the other hand, memory consumption soon becomes a problem as the kinetic energy and number of atoms to be considered increase. As an approximation, it is possible to only consider a single scattering from the emitter to any atom in the cluster. This approximation is extremely computationally fast and can give satisfactory results for adsorbates. We’ll see later that this approach is rather too simplistic for most cases.
Oxygen on Rh(001)#
In a paper published in 1998, T. Gerber et al. used the quite high backscattering factor of Rhodium atoms to probe the distance of Oxygen atoms adsorbed on a Rhodium surface. Some electrons coming from Oxygen atoms are ejected toward the Rhodium surface. They are then backscattered and interfere with the direct signal comming from Oxygen atoms (see the figure below). They demonstrated both experimentally and numerically with a sinle scattering computation that this lead to a very accurate probe of adsorbed species that can be sensitive to bond length changes of the order of \(\pm 0.02 \mathring{A}\).
See also
based on this paper from T. Greber et al. Phys. Rev. Lett. 81(8) p1654 (1998)
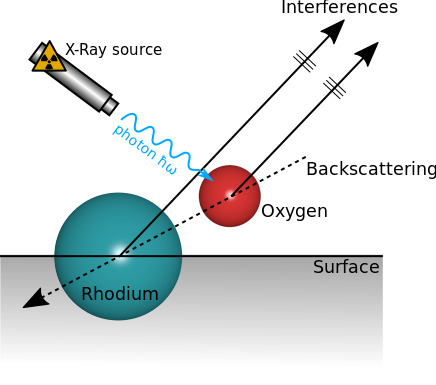
Fig. 6 Interferences produced by the backscattering effect#
Computing the scattering factor#
To illustrate that photoelectrons emitted by Oxygen adsorbates towards the Rhodium surface can be backscattered, we will start by computing the scattering factor for both O and Rh atoms.
By using the Atoms class of the ase package, try to build a O-Rh chain where atoms are 4 Å apart. Here is the begining of the script. Try to complete the line of code and view your two-atoms chain.
from ase import Atoms
from ase.visualize import view
# Create an atomic chain O-Rh
cluster = Atoms(... # Fill this line
Show code cell content
from ase import Atoms
from ase.visualize import view
# Create an atomic chain O-Rh
cluster = Atoms(['O', 'Rh'], positions = [(0,0,0), (0,0,4.)])
As previously, we create a calculator, we attach our 2 atoms cluster to this calculator and we define the first atom in the chain as the emitter
calc = MSSPEC(spectroscopy='PED')
calc.set_atoms(cluster)
cluster.emitter = 0
We use the get_scattering_factors method (see its documentation) to compute the scattering factors at 723 eV.
How large is the backscattering factor of Rhodium with respect to that of Oxygen ?
# Compute the scattering factor
data = calc.get_scattering_factors(kinetic_energy=723)
# Popup the results
data.view()
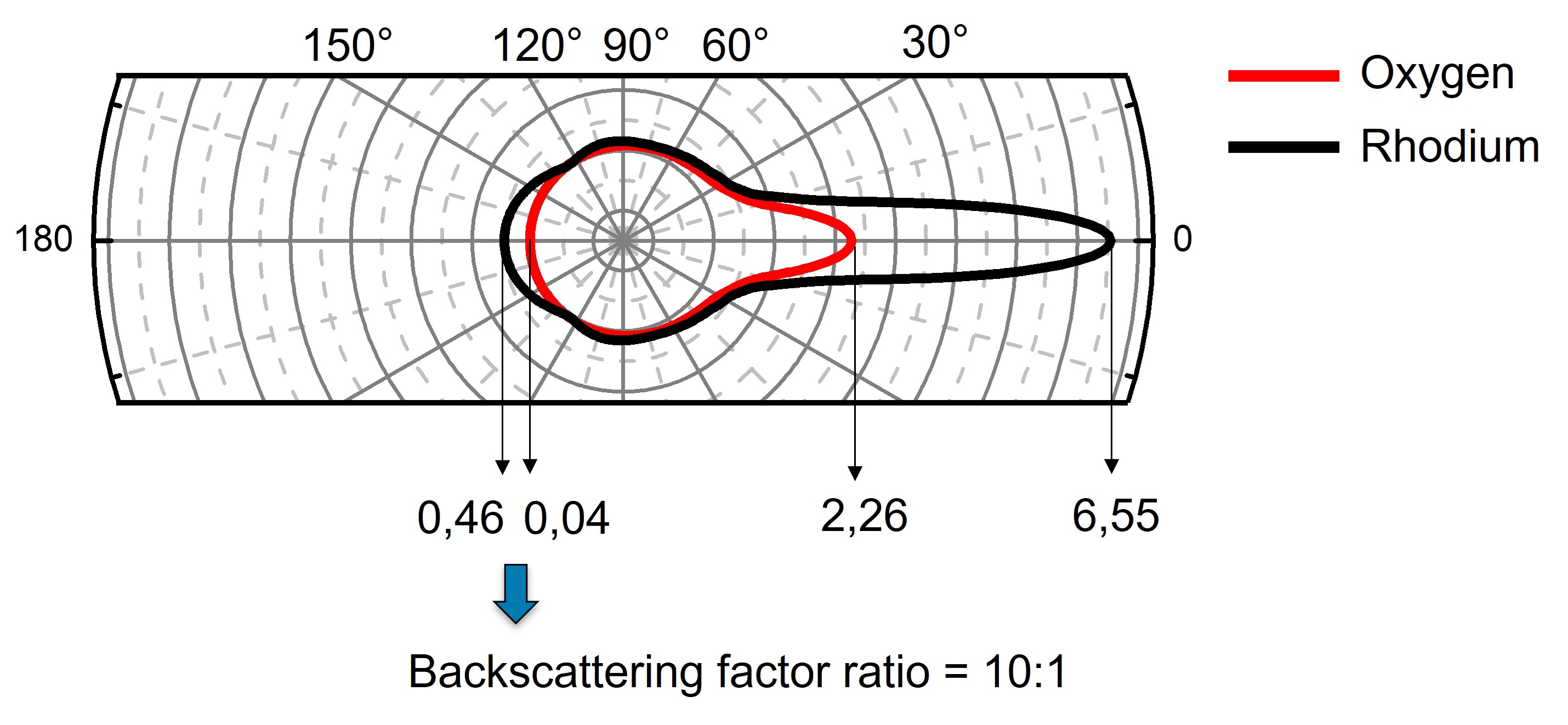
Fig. 7 Polar representation of the scattering factor#
Interferences due to backscattering#
Let an Oxygen atom (in red) being adsorbed at a distance \(z_0\) of an fcc site of the Rh(111) surface.
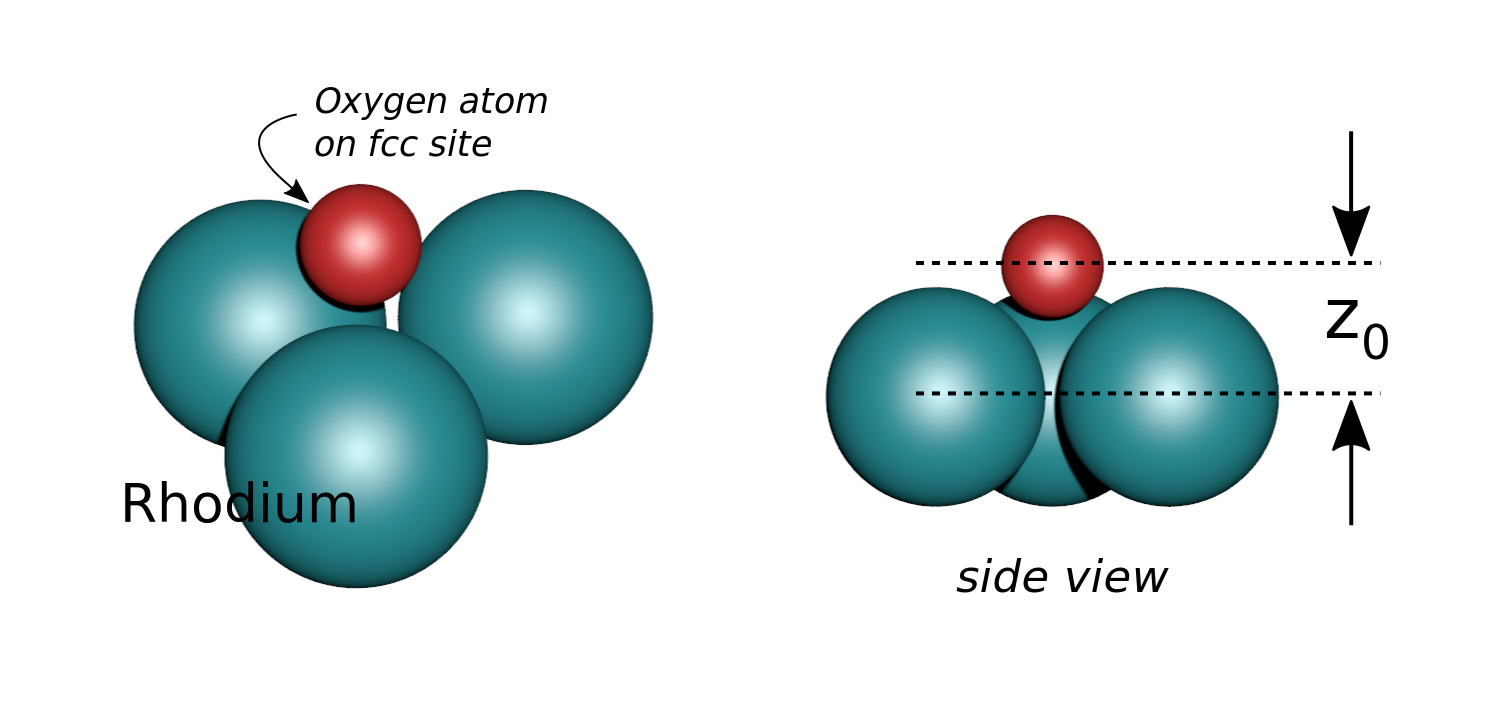
Fig. 8 Small cluster used for the calculation.#
We will compute for different values of the adsorption height \(z_0\).
Complete the script below to compute the (\(\theta,\phi\)) scan of the photodiffraction of O(1s) adsorbed on a fcc site on Rh(111) surface.
1from msspec.calculator import MSSPEC
2from ase.build import fcc111, add_adsorbate
3import numpy as np
4
5data = None
6all_z = ... # -> Define a list of z values for the adsorbate
7
8for ... # -> Complete this for-loop over z values
9 # construct the cluster
10 cluster = fcc111('Rh', size = (2,2,1))
11 cluster.pop(3)
12 add_adsorbate(... # -> Put the oxygen atom on the fcc site
13 cluster.emitter = ... # -> Oxygen is the last atom we added, so the indice is...
14
15 # Define a calculator for single scattering calculations
16 calc = MSSPEC(spectroscopy='PED', algorithm='expansion')
17 calc.calculation_parameters.scattering_order = 1
18 calc.set_atoms(cluster)
19
20 # Compute
21 data = calc.get_theta_phi_scan(level='1s', kinetic_energy=723, data=data)
22
23data.view()
As proposed in the comments, add a loop to vary the adsorption height of Oxygen between 1.10 and 1.65 Å. What is the bond length difference between to intensity maxima ?
1from msspec.calculator import MSSPEC
2from ase.build import fcc111, add_adsorbate
3import numpy as np
4
5data = None
6all_z = np.arange(1.10, 1.65, 0.05)
7
8for zi, z0 in enumerate(all_z):
9 # construct the cluster
10 cluster = fcc111('Rh', size = (2,2,1))
11 cluster.pop(3)
12 add_adsorbate(cluster, 'O', z0, position='fcc')
13 cluster.emitter = len(cluster) - 1
14
15 # Define a calculator for single scattering calculations
16 calc = MSSPEC(spectroscopy='PED', algorithm='expansion')
17 calc.calculation_parameters.scattering_order = 1
18 calc.set_atoms(cluster)
19
20 # Compute
21 data = calc.get_theta_phi_scan(level='1s', kinetic_energy=723, data=data)
22 dset = data[-1]
23 dset.title = "{:d}) z = {:.2f} angstroms".format(zi, z0)
24
25data.view()
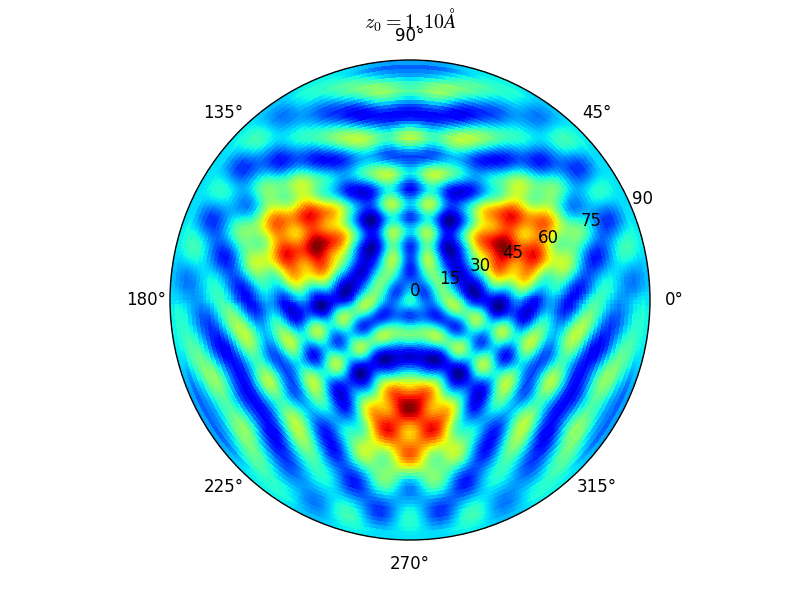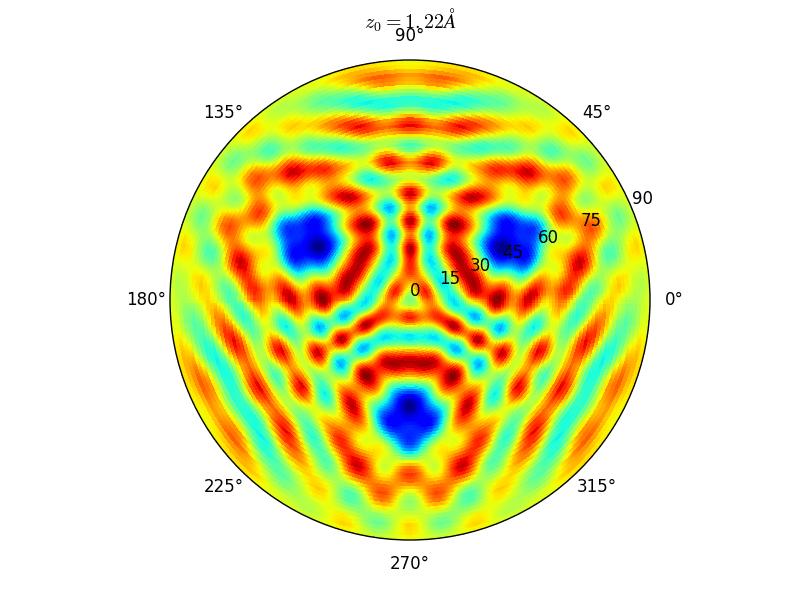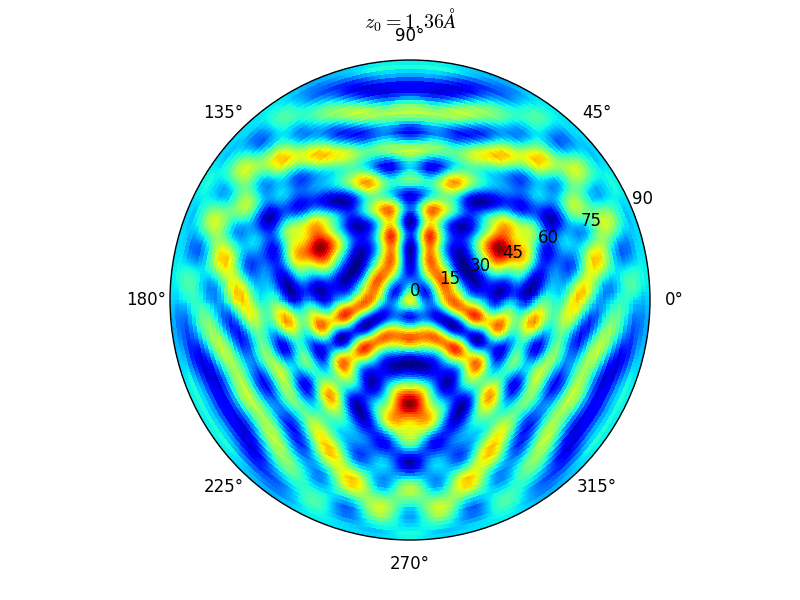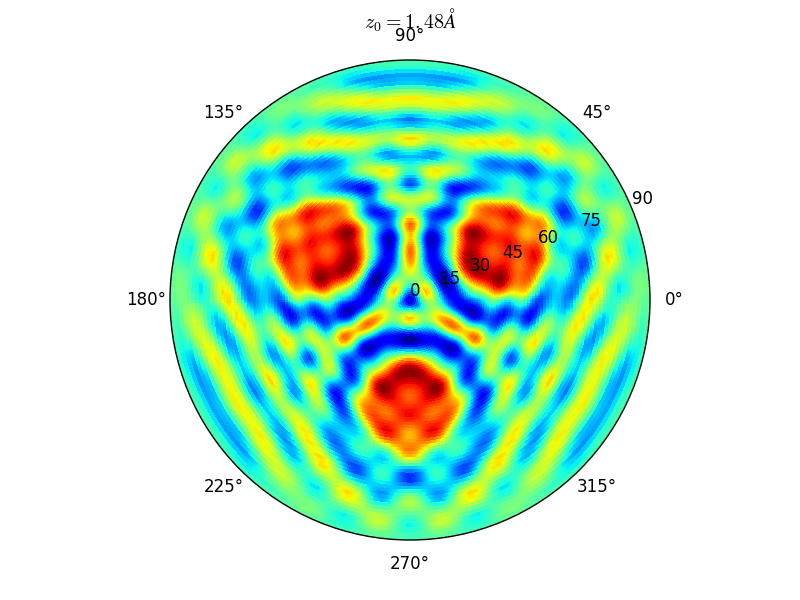
Fig. 9 Stereographic projections of O(1s) emission at \(E_0\) = 723 eV for an oxygen atom on top of a fcc site of 3 Rh atoms at various altitudes \(z_0\)#
You can see on the stereographic projection 3 bright circles representing fringes of constructive interference between the direct O(1s) photoelectron wave and that backscattered by the Rhodium atoms. The center of these annular shapes changes from bright to dark due to the variation of the Oxygen atom height above the surface which changes the path difference.