8.4 KiB
Your first XPD pattern
In this small example, you will learn how to produce a polar scan, ie you will plot the number of photo-electrons as a function of the θ angle. We will do this calculation for a bulk copper (001) surface.
This is a small script of roughly 15 lines (without comments) just to show you how to start with the msspec package. For this purpose we will detail all steps of this first hands on as much as possible.
Start your favorite text editor to write this Python script.
Begin by typing:
System Message: ERROR/3 (<stdin>, line 15)
Error in "code-block" directive: unknown option: "linenos".
.. code-block:: python
:linenos:
:lineno-start: 1
# coding: utf8
from msspec.calculator import MSSPEC
from msspec.utils import *
Every line starting by a '#' sign is considered as a comment in Python and thus is ignored... except the first line with the word 'coding' right after the '#' symbol. It allows you to specify the encoding of your text file. It is not mandatory but highly recommended. You will most likeley use an utf-8 encoding as in this example.
For an MsSpec calculation using ASE, msspec modules must be imported first. Thus, line 3 you import the MSSPEC calculator from the calculator module of the msspec package. MSSPEC is a class.
We will also need some extra stuff that we load from the utils module (line 4).
We need to create a bulk crystal of copper atoms. We call this a cluster. This is the job of the ase module, so load the module
System Message: ERROR/3 (<stdin>, line 41)
Error in "code-block" directive: unknown option: "linenos".
.. code-block:: python
:linenos:
:lineno-start: 6
from ase.build import bulk
from ase.visualize import view
a0 = 3.6 # The lattice parameter in angstroms
# Create the copper cubic cell
copper = bulk('Cu', a=a0, cubic=True)
view(copper)
In line 6 we load the :py:func:`bulk` function to create our atomic object and, in line 7, we load the :py:func:`view` function to actually view our cluster.
System Message: ERROR/3 (<stdin>, line 54); backlink
Unknown interpreted text role "py:func".System Message: ERROR/3 (<stdin>, line 54); backlink
Unknown interpreted text role "py:func".The creation of the cluster starts on line 12. We create first the copper cubic cell with the The :py:func:`bulk` function. It needs one argument which are the chemical species ('Cu' in our example). We also pass 2 keyword (optional) arguments here:
System Message: ERROR/3 (<stdin>, line 57); backlink
Unknown interpreted text role "py:func".
- The lattice parameter a in units of angströms.
- The cubic keyword, to obtain a cubic cell rather than the fully reduced one which is not cubic
From now on, you can save your script as 'Cu.py' and open a terminal window in the same directory as this file. Launch your script using 'python' or 'msspec -p' (depending on your installation):
System Message: WARNING/2 (<stdin>, line 70)
Cannot analyze code. Pygments package not found.
.. code-block:: bash
python Cu.py
or
System Message: WARNING/2 (<stdin>, line 76)
Cannot analyze code. Pygments package not found.
.. code-block:: bash
msspec -p Cu.py
and a graphical window (the ase-gui) should open with a cubic cell of copper like this one:
Figure 1.
Obviously, multiple scattering calculations need more atoms to be accurate. Due to the forward focusing effect in photo-electron diffraction, the best suited geometry for the cluster is hemispherical. Obtaining such a cluster is a straightforward process thanks to the :py:func:`utils.hemispherical_cluster` function. This function will basically create a cluster based on a pattern (the cubic copper cell here).
System Message: ERROR/3 (<stdin>, line 90); backlink
Unknown interpreted text role "py:func".Modify your script like this and run it again.
System Message: ERROR/3 (<stdin>, line 99)
Unknown directive type "literalinclude".
.. literalinclude:: Cu.py
:linenos:
:lineno-start: 1
:lines: 1-13
Don't forget to add the line to view the cluster at the end of the script and run the script again. The :py:func:`hemispherical_cluster` works in 3 simple steps:
System Message: ERROR/3 (<stdin>, line 105); backlink
Unknown interpreted text role "py:func".
- Repeat the given pattern in all 3 directions
- Center this new set of atoms and cut a sphere from the center
- Remove the upper half of the created 'sphere'.
To get more information about how to use this function, have a look at the :ref:`hemispherical_cluster_faq` section in the :ref:`faq`.
System Message: ERROR/3 (<stdin>, line 112); backlink
Unknown interpreted text role "ref".System Message: ERROR/3 (<stdin>, line 112); backlink
Unknown interpreted text role "ref".
Figure 2. The different steps to output a cluster. a) After repeat, b) after cutting a sphere, c) final cluster
Once your cluster is built the next step is to define which atom in the cluster will absorb the light. This atom is called the absorber (or the emitter since it emits the photoelectron).
To specify which atom is the absorber, you need to understand that the cluster object is like a list of atoms. Each member of this list is an atom with its own position. You need to locate the index of the atom in the list that you want it to be the absorber. Then, put that number in the cluster.absorber attribute
For example, suppose that you want the first atom of the list to be the absorber. You would write:
System Message: WARNING/2 (<stdin>, line 134)
Cannot analyze code. Pygments package not found.
.. code-block:: python
cluster.absorber = 0
To find what is the index of the atom you'd like to be the absorber, you can either get it while you are visualizing the structure within the ase-gui program (select an atom with the left mouse button and look at its index in the status line). Or, you can use :py:func:`utils.get_atom_index` function. This function takes 4 arguments: the cluster to look the index for, and the x, y and z coordinates. It will return the index of the closest atom to these coordinates. In our example, as we used the :py:func:`utils.hemispherical_cluster` function to create our cluster, the emitter (absorber) is always located at the origin, so defining it is straightforward:
System Message: ERROR/3 (<stdin>, line 138); backlink
Unknown interpreted text role "py:func".System Message: ERROR/3 (<stdin>, line 138); backlink
Unknown interpreted text role "py:func".System Message: ERROR/3 (<stdin>, line 148)
Unknown directive type "literalinclude".
.. literalinclude:: Cu.py
:linenos:
:lineno-start: 15
:lines: 15-16
That's all for the cluster part. We now need to create a calculator for that object. This is a 2 lines procedure:
System Message: ERROR/3 (<stdin>, line 157)
Unknown directive type "literalinclude".
.. literalinclude:: Cu.py
:linenos:
:lineno-start: 18
:lines: 18-22
When creating a new calculator, you must choose the kind of spectroscopy you will work with. In this example we choose 'PED' for PhotoElectron Diffraction. Other types of spectroscopies are:
- 'AED' for Auger Electron Spectroscopy
- 'APECS' for Auger PhotoElectron Coincidence Spectroscopy
- 'EXAFS' for Extended X-Ray Absorption Fine Structure
- 'LEED' for Low Energy Electron Diffraction
Now that everything is ready you can actually perform a calculation. The lines below will produce a polar scan of the Cu(2p3/2) level with default parameters, store the results in the data object and display it in a graphical window.
System Message: ERROR/3 (<stdin>, line 177)
Unknown directive type "literalinclude".
.. literalinclude:: Cu.py
:linenos:
:lineno-start: 24
:lines: 24-28
running this script will produce the following output
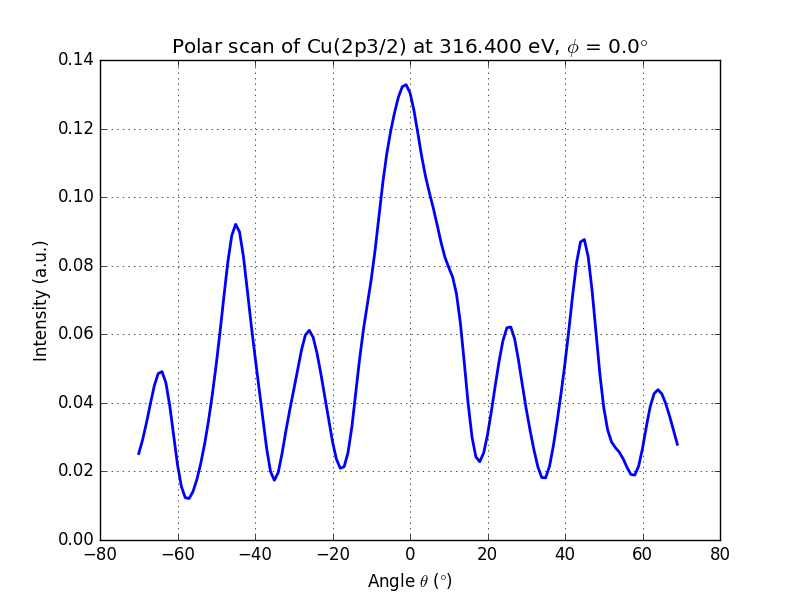
Figure 3. Polar scan of copper (2p3/2)
You can clearly see the modulations of the signal and the peak at θ = 0° and θ = 45°, which are dense directions of the crystal.
By default, the program computes for θ angles in the -70°..+70° range. This can be changed by using the angles keyword.
System Message: WARNING/2 (<stdin>, line 196)
Cannot analyze code. Pygments package not found.
.. code-block:: python
#For a polar scan between 0° and 60° with 100 points
import numpy as np
data = calc.get_theta_scan(level = '2p3/2', theta = np.linspace(0,60,100))
# For only 0° and 45°
data = calc.get_theta_scan(level = '2p3/2', theta = [0, 45])
You probably also noticed that we did not specify any kinetic energy for our photoelectron in this example. By default, the programm tries to find the binding energy (Eb) of the choosen level in a database and assume a kinetic energy (Ek) of
where ħω is the photon energy, an ΦWF is the work function of the sample, arbitrary set to 4.5 eV.
Of course, you can choose any kinetic energy you'd like:
System Message: WARNING/2 (<stdin>, line 219)
Cannot analyze code. Pygments package not found.
.. code-block:: python
# To set the kinetic energy...
data = calc.get_theta_scan(level = '2p3/2', kinetic_energy = 300)
Below is the full code of this example. You can download it :download:`here <Cu.py>`
System Message: ERROR/3 (<stdin>, line 226); backlink
Unknown interpreted text role "download".System Message: ERROR/3 (<stdin>, line 229)
Unknown directive type "literalinclude".
.. literalinclude:: Cu.py
:linenos:
System Message: ERROR/3 (<stdin>, line 232)
Unknown directive type "seealso".
.. seealso::
Atomic Simulation Environnment
`Documentation <https://wiki.fysik.dtu.dk/ase/python.html>`_
of the ASE module to create your clusters much more.
X-Ray data booklet
`Electron binding energies <http://xdb.lbl.gov/>`_ are
taken from here.